
定位显微镜
基于定位的超分辨率技术,如 STORM, dSTORM/GSDIM or (spt)PALM,都依赖于时间去相关的荧光。通过使单个分子发光分离,然后以亚衍射精度确定其位置,从而重建超高分辨率图像。为了实现这一目标,视野内的绝大多数分子必须处于非发光状态。
为了实现荧光发射的时间分离,开发了几种方法:
- STORM(随机光学重建显微镜)通过高功率激光照射,将大部分分子置于暗态,从而实现单个分子的时间分辨荧光。 [1, 2, 3]. 为了使这种方法有效,样本必须置于氧化或还原缓冲液中,以稳定荧光分子的关闭状态。随机地,一小部分分子会从暗态跃迁到发光态。这种“开启速率”可以通过改变周围溶液的条件和使用第二束激光(405 nm)来调节。
- PALM(光激活定位显微镜)技术则利用可光激活或转换的荧光蛋白,这些蛋白可以被“打开”和“关闭”。[4, 5]. 激活后,这些蛋白会持续发光直到淬灭。激活光源的强度决定了荧光蛋白的激活速率。
尽管定位显微技术已经实现了亚衍射分辨率,但要在活细胞中对天然蛋白进行超高分辨率成像仍然是一个挑战。
当研究人员需要在活细胞中研究高密度的内源性蛋白时,STORM和PALM技术都无法完全满足要求。 STORM方法所需的还原性成像缓冲液与活细胞不相容。而PALM则依赖于基因改造的荧光蛋白,因此无法观察内源性蛋白。
uPAINT(纳米尺度拓扑成像通用点积累)是一种巧妙的技术 [6]。它通过动态成像细胞表面单个分子的运动轨迹,实现了对活细胞表面天然生物分子行为的超高分辨率追踪。
与传统的定位显微技术不同,uPAINT通过成像低浓度的荧光配体与其靶标在小光体积内的结合和相互作用,实现了荧光信号的时间分离。
uPAINT 原理
uPAINT技术源于早期的PAINT(纳米尺度拓扑成像点积累)技术。在PAINT中,荧光标记的探针在溶液中扩散,一旦与目标结合就会发出荧光。Alexey Sharanov和Robin Hochstrasser率先使用尼罗红对脂质双层和大型单层囊泡进行连续标记,实现了这一方法。尼罗红在水溶液中不发光,但在疏水环境中会发光[7]。水分子快速扩散,使得尼罗红在膜附近持续存在。一旦进入膜,探针就会发出荧光,其移动性显著降低,从而可以通过相机检测到(持续数毫秒),直到发生光漂白。通过确定每个荧光信号的质心,可以达到约25纳米的分辨率。然而,由于插入时间极短(毫秒级),无法长时间追踪单个分子的行为。
随后,Gregory Giannone和Eric Hosy着重于克服原始PAINT技术的某些局限性。通过使用特异性荧光配体(如抗体)替代非特异性膜染料,实现了对单个特定蛋白质相互作用的动态成像。由于应用范围更广,这种方法被命名为通用PAINT [6].
对于该技术,通过倾斜照明(如图1所示)克服了介质中未结合标记配体的背景荧光。关键是将TIRF激光从其原有用途转变。将激光入射角略低于临界角,使得光束在载玻片上形成一个“高度倾斜层状光学片”(HILO),斜穿过样品。只有在这个光学片内的荧光分子才会被激发。
将低浓度的标记配体添加到样品周围的缓冲液中。布朗运动使得配体持续随机地与细胞表面的靶标结合。只有在HILO光束路径内的荧光分子才会被激发并成像(如图1所示)。


图 1:uPAINT 原理。A) 徕卡 SR GSD 3D 提供了调整激光束穿透角度的机会。通过它可以实现TIRF(全内反射荧光)照明,前提是激光击中玻璃载玻片-样本界面超出所谓的临界角θ
与PAINT中使用的脂质探针的荧光信号主要受插入时间影响且持续时间较短不同,uPAINT中使用的配体信号主要受荧光淬灭的影响。
因此,通过uPAINT,我们可以以超高分辨率追踪并记录结合配子的运动轨迹。对这些轨迹进行分析和重建,可以获得分子运动的轨迹信息。由于能够收集到大量数据,我们可以对配子的运动速度、扩散范围、聚集状态等进行定量分析。例如,我们可以利用uPAINT研究突触处膜受体的动态行为(如图2所示)。
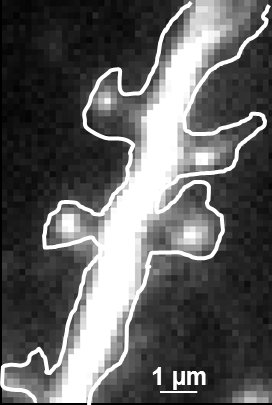



图 2:uPAINT 图像形成过程。A) 转染了突触标记蛋白Homer-GFP的神经元荧光显微图像(用于勾勒细胞轮廓)。B) 实验获取的单个图像示例。四个被良好分离的单个荧光配体(ATTO 647)与目标结构结合。每个粒子通过计算进行超定位(像素大小160 nm)。C) 所有单个粒子定位的叠加。亮像素对应于树突中被频繁访问的区域(像素大小20 nm)。D) 单个蛋白质的运动轨迹。不同颜色代表不同粒子的运动轨迹。超分辨率图像是使用 JB Sibarita 团队(波尔多大学)开发的软件获得的,商业上可用作 WaveTracer MetaMorph 插件。
各种生物学问题都可以通过uPAINT技术来探索。因此,需要根据不同的研究目的仔细选择荧光探针。例如,对于需要到达细胞内受限区域的情况,可以使用尺寸较小的探针,如TrisNTA-atto或Fab片段(约2nm [6]). 而对于需要记录长时程的分子运动轨迹,多重偶联抗体则更为合适。有趣的是,甚至可以开发单分子FRET实验[8].
总之,uPAINT不仅能生成超分辨率图像,还能提供活细胞中特定膜结合生物分子在单分子水平上的动态信息。与传统的定位显微镜技术相比,uPAINT无需使用有害的缓冲液或进行基因改造,因此可以在接近生理条件下进行观察[9].
uPAINT技术的一个局限性在于它主要用于研究质膜上的分子。由于荧光标记的抗体无法穿透活细胞的完整质膜,因此无法利用uPAINT来研究细胞内的蛋白质动态。
使用 Leica SR GSD 3D 的 uPAINT
徕卡SR GSD 3D显微镜提供了两种基本操作模式:表征荧光和全内反射荧光(TIRF)照明。对于TIRF照明,TIRF照明器将激光束准直并聚焦到覆盖玻片上,其中包含置于水溶液中的样品。当激光束入射到玻璃-水界面时,如果入射角超过临界角,就会发生全反射现象。同时,在玻璃滑片另一侧的样品中会产生一个衰减波。通过倾斜激光束入射角,可以调节衰减场的穿透深度,范围从80到250纳米。只有位于衰减场内的荧光分子会被激发,从而获得非常高的信噪比。因此,TIRF照明是观察质膜附近过程的首选技术(图1)。
通过将激光入射角降低至临界角以下,激光束便能穿过载玻片,并以倾斜角度照射样品。这意味着只有样品的一小部分会被激发光照亮。这种被称为HILO(高度倾斜层状光学片)的照明模式下获取的图像具有低背景和高信噪比的特点。此外,该方法还可以减少光漂白和光毒性,从而延长细胞的存活时间(图1)。
在实际操作中,徕卡SR GSD 3D显微镜提供了两种不同的工作模式来控制激光束,分别用于TIRF和HILO照明(见图3)。
自动模式允许用户在80到250纳米的范围内,以步进的方式选择预设的穿透深度。此外,还可以通过四种不同的方式来确定衰减场的方向(方位角)。
图3:自动模式与专家模式:激发激光束的穿透深度和方位可以通过成像软件进行控制。A) 自动模式允许逐步选择从 80 到 250 纳米的消逝场穿透深度,以及四个不同的消逝场方向。B) 专家模式允许使用滑块无级设置穿透深度。绿色区域标记了TIRF区域。C) 将滑块移出绿色TIRF区域可实现 HILO 照明。
对于uPAINT实验,将生长在盖玻片上的细胞用非荧光培养液(如Tyrode's培养液)覆盖,然后放置在显微镜上。在明场模式下找到感兴趣的区域并聚焦后,切换到GSD模式,选择正确的滤光片和激光。接下来,利用专家模式找到合适的HILO照明设置(如上所述)。需要注意的是,滑块的位置以及激光入射角取决于细胞的厚度。随后,添加荧光标记的抗体或配体到培养液中。在随后的采集过程中,只有结合在质膜上的抗原的荧光分子会被激发。由于斜照明,未结合的配体不会被激发,从而减少了漂白并降低了噪声水平。在实验过程中,可以持续对质膜蛋白的标记进行成像。原始数据文件随后可用于重建单个分子的定位和动态信息,实现超分辨率成像。
uPAINT 实验有时需要较长的采集时间(数十秒)来揭示几乎所有感兴趣蛋白质的运动和组织。在这种情况下,可以通过图像到图像检测珠子的位置来校正漂移(例如,TetraSpeckTM)。采集后,Leica SR GSD 3D 软件能够根据珠子的定位自动重新定位每个图像。这种漂移校正模式在研究纳米尺度聚集体时是必不可少的。
总之,Leica SR GSD 3D 提供了不同的照明选项。其原始用途是在 dSTORM/GSDIM 超分辨率显微镜下进行 TIRF 或表面荧光照明。此外,还可以进行 HILO 照明,这对于 uPAINT 是必要的。因此,可以使用一台显微镜收集静态和动态的蛋白质数据,从而实现对活细胞内源水平的蛋白质组织和动态的超分辨率检查。
References
- Rust MJ, Bates M, and Zhuang X: Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3 (10): 793–96 (2006).
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, and Hell SW: Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 5 (11): 943–45 (2008).
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, and Sauer M: Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47 (33): 6172–76 (2008).
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, and Hess HF: Imaging intracellular fluorescent proteins at nanometer resolution. Science 313 (5793): 1642–45 (2006).
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-and Schwartz J: High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 5 (2): 155–57 (2008).
- Giannone G, Hosy E, Levet F, Constals A, Schulze K, Sobolevsky AI, Rosconi MP, Gouaux E, Tampé R, Choquet D, and Cognet L: Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 99 (4): 1303–10 (2010).
- Sharonov A, and Hochstrasser R: Wide-field subdiffraction imaging by accumulated binding of diffusing probes. PNAS 50: 18911–16 (2006).
- Winckler P, Lartigue L, Giannone G, De Giorgi F, Ichas F, Sibarita JB, Lounis B, and Cognet L: Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Sci. Rep. (2013).
- Zimmermann T, Hosy E: Webinar: New Dimensions in Super-Resolution Microscopy. Leica Science Lab (2014).
相关文章
-

Mica: 助力伦敦帝国学院开展跨学科科研研究
这篇访谈重点介绍了伦敦帝国学院的 Mica 所产生的变革性影响。科学家们解释了Mica如何改变了游戏规则,扩大了研究的可能性,促进了跨学科合作。他们解释了使用 Mica…
Feb 10, 2025Read article -

How to Study Gene Regulatory Networks in Embryonic Development
Join Dr. Andrea Boni by attending this on-demand webinar to explore how light-sheet microscopy…
Nov 20, 2024Read article -

